CAZypedia needs your help! We have many unassigned GH, PL, CE, AA, GT, and CBM pages in need of Authors and Responsible Curators.
Scientists at all career stages, including students, are welcome to contribute to CAZypedia. Read more here, and in the 10th anniversary article in Glycobiology.
New to the CAZy classification? Read this first.
*
Consider attending the 15th Carbohydrate Bioengineering Meeting in Ghent, 5-8 May 2024.
Glycosyltransferases
This page has been approved by the Responsible Curator as essentially complete. CAZypedia is a living document, so further improvement of this page is still possible. If you would like to suggest an addition or correction, please contact the page's Responsible Curator directly by e-mail.
- Author: Spencer Williams
- Responsible Curator: Spencer Williams
Overview
Glycosyltransferases are enzymes that catalyze the formation of the glycosidic linkage to form a glycoside. These enzymes utilize 'activated' sugar phosphates as glycosyl donors, and catalyze glycosyl group transfer to a nucleophilic group, usually an alcohol. The product of glycosyl transfer may be an O-, N-, S-, or C-glycoside; the glycoside may be part of a monosaccharide, oligosaccharide, or polysaccharide [1, 2, 3, 4, 5].
Glycosyl donor substrates
Glycosyltransferases can utilize a range of donor substrates. Sugar mono- or diphosphonucleotides are sometimes termed Leloir donors (after Nobel prize winner, Luis Leloir); the corresponding enzymes are termed Leloir glycosyltransferases.

Glycosyltransferases that utilize non-nucleotide donors, which may be polyprenol pyrophosphates, polyprenol phosphates, sugar-1-phosphates, or sugar-1-pyrophosphates, are termed non-Leloir glycosyltransferases.

In the last two cases, the enzymes that catalyze the transfer of a glycosyl group from a glycosyl phosphate or pyrophosphate are more commonly referred to as phosphorylases and pyrophosphorylases. Some of these enzymes are classified into glycoside hydrolase (GH) families (eg sucrose phosphorylase, GH13) and others are classified into GT familes (eg glycogen phosphorylase GT35).
For specific examples of glycosyl donor substrates, please see the Common sugar donors section below.
Mechanism
Glycosyltransferases catalyze the transfer of glycosyl groups to a nucleophilic acceptor with either retention or inversion of configuration at the anomeric centre. This allows the classification of glycosyltransferases as either retaining or inverting enzymes.
Inverting glycosyltransferases
Structural and kinetic data for inverting glycosyltransferases support a mechanism that proceeds through a single nucleophilic substitution step, facilitated by an enzymic general base catalyst. The transition state is believed to possess substantial oxocarbenium ion character. Most inverting glycosyltransferases require a divalent cation (typically Mg2+ or Mn2+) although metal-independent enzymes are known [2].

Retaining glycosyltransferases
Mechanistic evidence for the catalytic process that results in retention of configuration of glycosyltransferases is scant. One mechanism that seems reasonable by comparison with the classical Koshland retaining mechanism for glycoside hydrolases, involves an enzymic nucleophile. In this mechanism, the enzymic nucleophile reacts with the glycosyl donor to generate a glycosyl enzyme with inversion of stereochemistry, followed by reaction with the glycosyl acceptor with a second inversion of stereochemistry, to yield a product glycoside with a net retention of anomeric stereochemistry. However, attempts to trap a glycosyl enzyme on a wildtype glycosyltransferase have met with universal failure.
Further, crystal structures of retaining glycosyltransferases are mostly ambiguous with respect to identifying possible candidate nucleophiles, with some lacking any suitable nucleophile situated close enough to the glycosyl donor to be able to form a covalent intermediate. In other cases structural analysis has identified possible candidate nucleophiles, which mutagenesis studies have supported. In some cases mutagenesis of the candidate residue to a non-nucleophilic amino acid has not resulted in the expected, dramatic loss in catalytic activity (of the order of 106), as seen for retaining glycoside hydrolases.
The most common alternative mechanism that is invoked is one that involves an SNi process, also termed 'internal return'. In this process, the leaving group on the donor departs, and the nucleophile attacks from the same face, with the other face of the donor being blocked by the enzyme. An open question is whether this process is concerted (i.e. involving a single transition state in which bonds are formed and broken at similar times) or stepwise (i.e. involving two transition states, and therefore an oxocarbenium ion intermediate). A detailed kinetic study of the retaining glycosyltransferase OtsA (which catalyzes the synthesis of trehalose-6-phosphate from UDP-glucose and glucose-6-phosphate) yielded data that was consistent with a frontside, SNi-type reaction [6].

Classification
Sequence based classification
Sequence-based classification uses algorithmic methods to assign protein sequences to various families. The glycosyltransferases have been classified into more than 90 families [7, 8]. Each family (GT family) contains proteins that are related by sequence, and by corollary, fold. This allows several useful predictions to be made since the catalytic machinery is conserved within each family. Usually, the mechanism used (i.e. retaining or inverting) is also conserved within a GT family. An actively curated list of GT families and members is available through the Carbohydrate Active enZyme database [9].
3-D folds
In striking contrast to glycoside hydrolases, which exhibit a wide variety of folds, GTs exhibit a much narrower range of folds.
Leloir GTs
Sugar nucleotide-dependent (Leloir) glycosyltransferases have been found to possess only two different folds, termed the GT-A and GT-B folds [10]. The GT-A fold is typified by the first member to have its X-ray structure determined, SpsA fromBacillus subtilus [11]. The GT-A fold consists of two dissimilar domains, one involved in nucleotide binding and the other binding the acceptor. The GT-B fold was exemplified by its first member, the beta-glucosyltransferase from bacteriophage T4 [12]. The GT-B fold consists of two similar Rossmann fold subdomains.
Non-Leloir GTs
Non-Leloir glycosyltransferases possess non-GT-A and -B folds. For example, the polymerizing glycosyltransferase transglycosylase, which catalyzes the condensation of oligosaccharyl polyprenolphosphate to generate the carbohydrate backbone of peptidoglycan has a bacteriophage-lysozyme-like fold [13]. The Pyrococcus furiosius oligosaccharyltransferase STT3, which catalyzes the transfer of a preformed oligosaccharide from a dolichol phosphate glycosyl donor to form asparagine-linked glycoproteins, possesses a novel fold; however, this crystallized fragment is catalytically-inactive and may not represent the structure of the complete glycosyltransferase [14]. The same fold is also observed in the periplasmic domain of the PglB oligosaccharyltransferase of Camplyobacter lari; this latter protein was been crystallized as a full length protein incorporating a transmembrane domain that retains transferase activity [15]. A structure of PglB oligosaccharyltransferase in complex with a hexapeptide substrate showed that the peptide binds to both the periplasmic domain and the transmembrane domain. These data demonstrate that the transmembrane domain of this bacterial oligosaccharyltransferase is required for both substrate binding and catalysis.
Role of metals
Many, but not all, glycosyltransferases utilize divalent metal ion cofactors such as Mn2+ and Mg2+. These metals are found mainly in glycosyltransferases that are diphosphonucleoside-dependent. X-ray crystallographic analysis reveals that the metal ion is coordinated to an oxygen of each of the two phosphate groups, as well as to side-chain carboxylates derived from the protein. Much has been made of the so-called ‘DXD’ amino acid sequence as an identifier of glycosyltransferases, where the aspartate residues of this sequence are presumed to comprise the metal-binding residues of the active site. However, it is to be cautioned that no part of the DXD motif is invariant among glycosyltransferases, with this motif being present in more than 50% of all protein sequences. Moreover, it is emphasized that many glycosyltransferases are metal-ion independent, and thus do not bind metals at the active site, and so do not require a metal-binding motif at their active site.
Common sugar donors
- Examples of sugar nucleotide donors
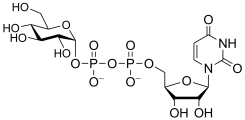
uridine diphospho-D-glucose
(UDP-Glc)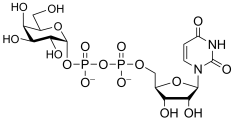
uridine diphospho-D-galactose
(UDP-Gal)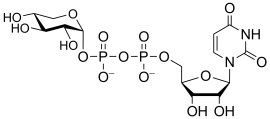
uridine diphospho-D-xylose
(UDP-Xyl)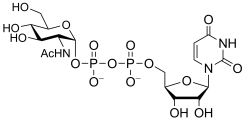
uridine diphospho-N-acetyl-D-glucosamine
(UDP-GlcNAc)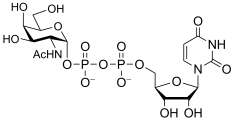
uridine diphospho-N-acetyl-D-galactosamine
(UDP-GalNAc)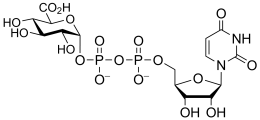
uridine diphospho-D-glucuronic acid
(UDP-GlcA)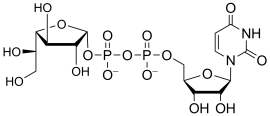
uridine diphospho-D-galactofuranose
(UDP-Galf)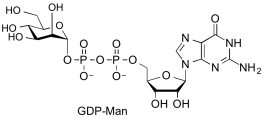
guanosine diphospho-D-mannose
(GDP-Man)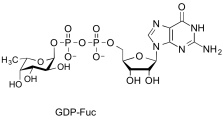
guanosine diphospho-L-fucose
(GDP-Fuc)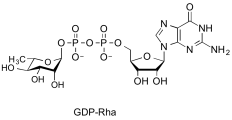
guanosine diphospho-L-rhamnose
(GDP-Rha)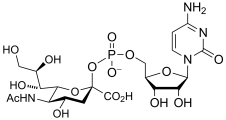
cytidine monophospho-N-acetylneuraminic acid
(CMP-Neu5Ac)
cytidine monophospho-2-keto-3-deoxy-D-mannooctanoic acid
(CMP-Kdo)












- Examples of sugar phospholipid donors
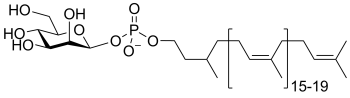
dolichol phosphomannose
(DP-Man)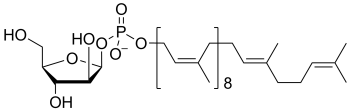
decaprenolphosphoarabinose
(PP-Ara)


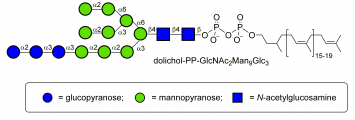
dolichol-PP-Glc3Man9GlcNac2

References
- Robert V. Stick and Spencer J. Williams. (2009) Carbohydrates. Elsevier Science.
- Lairson LL, Henrissat B, Davies GJ, and Withers SG. (2008). Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521-55. DOI:10.1146/annurev.biochem.76.061005.092322 |
- Coutinho PM, Deleury E, Davies GJ, and Henrissat B. (2003). An evolving hierarchical family classification for glycosyltransferases. J Mol Biol. 2003;328(2):307-17. DOI:10.1016/s0022-2836(03)00307-3 |
- Campbell JA, Davies GJ, Bulone V, and Henrissat B. (1997). A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem J. 1997;326 ( Pt 3)(Pt 3):929-39. DOI:10.1042/bj3260929u |
- Claus-Wilhelm von der Lieth, Thomas Luetteke, and Martin Frank. (2010-01-19) Bioinformatics for Glycobiology and Glycomics: An Introduction. Wiley.
Chapter 5: Coutinho PM, Rancurel C, Stam M, Bernard T, Couto FM, Danchin EGJ, Henrissat B. "Carbohydrate-active Enzymes Database: Principles and Classification of Glycosyltransferases."
- Lee SS, Hong SY, Errey JC, Izumi A, Davies GJ, and Davis BG. (2011). Mechanistic evidence for a front-side, SNi-type reaction in a retaining glycosyltransferase. Nat Chem Biol. 2011;7(9):631-8. DOI:10.1038/nchembio.628 |
-
Carbohydrate Active Enzymes database; URL http://www.cazy.org/
- Unligil UM and Rini JM. (2000). Glycosyltransferase structure and mechanism. Curr Opin Struct Biol. 2000;10(5):510-7. DOI:10.1016/s0959-440x(00)00124-x |
- Charnock SJ and Davies GJ. (1999). Structure of the nucleotide-diphospho-sugar transferase, SpsA from Bacillus subtilis, in native and nucleotide-complexed forms. Biochemistry. 1999;38(20):6380-5. DOI:10.1021/bi990270y |
- Vrielink A, Rüger W, Driessen HP, and Freemont PS. (1994). Crystal structure of the DNA modifying enzyme beta-glucosyltransferase in the presence and absence of the substrate uridine diphosphoglucose. EMBO J. 1994;13(15):3413-22. DOI:10.1002/j.1460-2075.1994.tb06646.x |
- Lovering AL, de Castro LH, Lim D, and Strynadka NC. (2007). Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science. 2007;315(5817):1402-5. DOI:10.1126/science.1136611 |
- Igura M, Maita N, Kamishikiryo J, Yamada M, Obita T, Maenaka K, and Kohda D. (2008). Structure-guided identification of a new catalytic motif of oligosaccharyltransferase. EMBO J. 2008;27(1):234-43. DOI:10.1038/sj.emboj.7601940 |
- Lizak C, Gerber S, Numao S, Aebi M, and Locher KP. (2011). X-ray structure of a bacterial oligosaccharyltransferase. Nature. 2011;474(7351):350-5. DOI:10.1038/nature10151 |